 Néhány nappal ezelőtt már közzétettük a Corona_Fakten Telegram-csatornánkon kontrollkísérletünk 1. fázisát, amelyben megvitattuk az általunk megbízott laboratórium eredményeit az úgynevezett citopátiás hatásról. [Közzétevő: A cikk tartalma részben azonos az általunk lefordított
Néhány nappal ezelőtt már közzétettük a Corona_Fakten Telegram-csatornánkon kontrollkísérletünk 1. fázisát, amelyben megvitattuk az általunk megbízott laboratórium eredményeit az úgynevezett citopátiás hatásról. [Közzétevő: A cikk tartalma részben azonos az általunk lefordított
Az általánosan elfogadott víruselmélet cáfolata – Alapok c. cikkel]
A kapott eredmények egyértelműen bizonyítják, hogy ez a hatás nem állítható be a vírus jelenlétének bizonyítékaként – ahogyan azt a virológusok 1954 óta állítják – hanem maga a kísérleti elrendezés váltja ki a hatást, amely akkor is fellép, ha nem adunk a kultúrához állítólagosan fertőzött anyagot.
Ha valaki még nem ismerné ezeket a kontrollkísérleteket, az a [14] hivatkozásnál részletesen tanulmányozhatja őket.
Ma bemutatjuk a folytatást: a kontrollkísérletek 2. fázisát
Gyakran előfordul, hogy egyetlen tanulmány szolgál alapul az összes többi tudós további kutatásának, és ez a SARS-CoV-2 esetében sincs másképp.
Nem nehéz azonban elképzelni a messzemenő következményeket, ha éppen ez a tanulmány – amely ebben az esetben világszerte mindenki más elé tárja, hogyan kell az állítólagos betegséget okozó vírusnak genetikailag kell kinéznie – tudományellenes megközelítéssel hamis és téves adatokat hozott létre.
Nyilvánvalónak kellene lennie, hogy ez a téves tudományos következtetések lavináját indítja el; a jól ismert következményekkel, amelyeknek az elmúlt két évben a szemünk előtt zajlottak le.
Míg a kanyaróvírus esetében John F. Enders 1954-es tanulmánya volt az, amely minden további tanulmány „anyja” lett, addig a SARS-CoV-2 esetében központi szerepet kap Prof. Zhang et. al. tudományos tanulmánya, amely a „Nature”-ben jelent meg és a következő címet viseli:
„A new coronavirus associated with human respiratory disease in China” = Egy új koronavírus Kínában, emberi légzőszervi megbetegedésekkel összefüggésben „[1] .
Miután a legapróbb részletekig ízeire szedtük Prof. Zhang et. al. tanulmányát, megdöbbenve kellett magunknak beismernünk, hogy a kínai tudósok nemcsak, hogy súlyos hiányosságokon siklottak át, de még a nyílt hamisítást is elfogadható eszköznek tekintették. Mi más ez, ha nem csalás?
- Az összes közzétett szekvencia fele ki van feketítve, így az eredeti nukleotid-sorrend már nem áll rendelkezésre. Így a megismételhetőség, a rekonstruálás lehetetlenné válik a tudósok számára. [13]
- Egyetlen vírust sem sikerült izolálni, még csak nem is centrifugálták, nemhogy ülepítettek volna.
- A SARS-CoV-2 genomjának összeállítása (assembly), amely világszerte minden tudós számára mintául szolgált, nem reprodukálható a kínaiak közzétett szekvenciáival.
- Nincs bizonyíték arra, hogy az állítólagos SARS-CoV-2 genomot tiszta vírus-RNS-ből állították volna elő.
- A kínai szerzőket finoman befolyásolhatta, akár öntudatlanul is, hogy a vizsgált beteg kórtörténetének és tüneteinek ismeretében csak a lehetséges légúti kórokozók keresésére koncentráltak.
- A kínaiak két teljesen eltérő végeredményt kaptak. A „Megahit” programmal átfedéssel talált leghosszabb összefüggő szekvencia 30 474 nukleotid hosszúságú volt, míg a másik program, a „Trinity” 11 760 nukleotid hosszúságú szekvenciát alkotott ugyanabból az adathalmazból. Ezzel szemben a Trinity lényegesen több egybefüggő szekvencia-darabot, nevezetesen 1 329 960 darabot állított elő, mint a Megahit (384 096). Ez tudományos szempontból rendkívül kérdéses, főleg a reprodukálhatóság szempontjából.
- A nyilvánosság számára hozzáférhetővé tett kínai szekvencia-adatok nem az eredeti, nyers szekvencia-adatok.
- Nem adtak meg ok-okozati összefüggést az állítólagos vírus és a szóban forgó betegség között, a szerzők nem tudtak ilyen prezentálni. A megállapítás tehát csak rendkívül gyenge korrelációt jelent, mivel csak EGY beteget vizsgáltak!
- A szekvenciák valószínűleg emberi eredetűeket is tartalmaznak, pontosabban: riboszomális RNS-t (legalábbis, ha a virológusok adatbázisai helyesek). Eszerint az RNS-kiszűrés során nem az összes riboszómális RNS-t távolították el.
- A célgenom lefedettsége a kapcsolt rövid szekvenciákkal (PCR-primerek) nagyon egyenetlen eloszlást mutat. Az elfogulatlan (torzulás nélküli) szekvenálás esetén azt várnánk, hogy a talált szekvenciák nagyjából minden nukleotidot hasonló számúszor fednek le. A binomiális eloszlás egyszerűsített feltételezése alapján azt várnánk, hogy a lefedettség többnyire az ábrázolt folyosón belül helyezkedik el. A lefedettség jelentős eltérése felveti a kérdést, hogy a szekvenálási lépés során keletkeztek-e véletlenül olyan szekvenciák, amelyek nem voltak meg az eredeti mintában.
Nem végeztek kontrollkísérleteket.
Néhány példa a nélkülözhetetlen kontrollkísérletekre, amelyeket a virológusok nem végeztek el:
1, Kontrollkísérletek, amelyek kizárják annak lehetőségét, hogy a virológusok által a vírus kimutatásaként állított citopátiás hatás nem magának a kísérleti elrendezésnek köszönhető. Néhány tanulmányban azt állítják, hogy elvégezték ezt a kontrollt, de ezt soha nem dokumentálták, és ez lehetetlenné teszi az ellenőrzést
2, A másik, a tudományos logikából következő kontrollkísérlet a kifejlesztett PCR-eljárás (valós idejű RT-PCR) intenzív tesztelése a vírusnak tulajdonított betegségektől eltérő, más betegségekben szenvedő emberek klinikai mintáival, valamint egészséges emberekből, állatokból és növényekből származó mintákkal annak ellenőrzésére, hogy az ő mintáik nem bizonyulnak-e szintén „pozitívnak”.
3, Ellenőrző kísérletek elvégzése, hogy kizárják annak lehetőségét.
- hogy egy egészséges személy tüdőváladékából származó humán/mikrobiális RNS mintából,
- egy másik tüdőbetegségben szenvedő személyből származó mintából,
- olyan személyből származó mintákból, akinek a SARS-CoV-2 tesztje negatív,
- vagy a SARS-CoV-2 vírus fölfedezése előtt vett és eltett mintákból származó RNS-ből
pontosan ugyanez a vírusgenom összeállítása lehetséges rövid RNS-töredékekből!
4, A „fertőzött sejtkultúrából” származó genomszekvenálásra vonatkozó kontrollkísérletek annak kizárására, hogy más vírusgenomokat is össze lehet-e állítani „de novo” assembler programmal (assembly) vagy más referencia genomokkal való rendezéssel (alignment).
5, Kontrollkísérletek annak kizárására, hogy a célvírus genomját elő lehet-e állítani negatív kontrollkultúrából „de novo” assembler folyamattal vagy rendezéssel (alignment).
Az igazi tudós, ezen a ponton egyszerűen megdöbben. Hát csakugyan lehetséges, hogy megszegték és figyelmen kívül hagyták az összes tudományos szabályt?
Meg tudja nekem valaki magyarázni, hogy miért nem vette észre a hatalmas hibákat egyetlen tudós sem, aki a kínaiak munkájára alapozta publikációját? Vagy egyszerűen csak példátlan vakmerőség kellett a tudománytalan munkához? Vagy egyszerűen csak tanácsos volt nekik a kínaiak hibáit nem észrevenni?
________________________________________
Új, emberi légúti megbetegedéseket okozó koronavírus Kínában – Csakugyan „új” és „betegséget okozó” vírust találtak Vuhanban?
1. rész: Hogyan határozták meg az „új” SARS-CoV-2 koronavírus szekvenciáját?
2. rész: A módszerek és következtetések kritikai áttekintése
Bevezetés
A jelenlegi világjárványban a kutató orvosok, elsősorban a virológusok kiemelkedő szerepet töltenek be. Értékeléseiket társadalmunkban alig kérdőjelezik meg, és a politika azokra támaszkodva hozza meg intézkedéseit. Úgy tűnik – különösen a társadalomra gyakorolt óriási hatást figyelembe véve – ez a megközelítés nem megfelelő. Ha mint társadalom, jelentős teret nyitunk a technokrata gondolkodás és cselekvés számára a társadalmi élet alakítása érdekében, akkor a szükséges ismeretekkel és a megfelelő módszertani készségekkel is rendelkeznünk kell mindenütt, különösen a döntéshozók soraiban. Ellenkező esetben a hit elsőbbséget élvez a tudással szemben. Ez egy olyan probléma, amely jelenleg drámai módon tárul fel, és rendkívül toxikussá válhat. Itt az ideje, hogy nagyon sok ember nagykorúvá váljon, elinduljon a kutatás útján, és bízzon saját képességeiben. Nem bizalmatlanságról van szó, hanem a megkérdőjelezésről. Az abszolút igazságot nem érhetjük el, és nem is várhatjuk el, hogy kétséget kizáróan létezzen. A tudást folyamatosan meg kell kérdőjelezni, tesztelni kell, és a dogmákat le kell leplezni. Ezeknek kell jellemezniük a tudományos munkát. Az új ötletekkel, érvekkel és a vélt ismeretek alternatív értelmezésével szemben mindig nyitottnak kell lennünk. mert ezek mint gyógyelixír gazdagítják életünket.
„Merészebb lehet kételkedni az ismertben, mint felfedezni az ismeretlent”. (Alexander von Humboldt)
Jelen cikkünk részletesen tárgyalja „A new coronavirus associated with human respiratory disease in China” [1] című tudományos közleményt, amelyben először javasolták az állítólagos „új” SARS-CoV-2 koronavírus genetikai szekvenciáját. A tanulmány 2020. február 03-án jelent meg a Nature tudományos folyóiratban.
Elemzésünk két részből áll. Az első részben ismertetjük az állítólagos SARS-CoV-2 vírusgenom létrehozásának módszertani eljárását. Ezt követi a második részben a kiadvány tudományos értelemben vett kritikai elemzése. Így olyan módszertani gyengeségekre, kritikus kérdésekre és ellenőrzési lehetőségekre hívjuk fel a figyelmet, amelyek ellenőrizhető módon megkérdőjelezik az újszerű és betegséget okozó vírus fölfedezését.
1. rész: Hogyan határozták meg az „új” SARS-CoV-2 koronavírus szekvenciáját?
A kiindulási helyzet
Egy 41 éves férfi beteget vizsgáltak, akit 2019. december 26-án vettek fel a Vuhani Központi Kórházba. A férfi a helyi halpiacon dolgozott, amelyet a járványügyi vizsgálatok az „újszerű” vírus eredeti kitörési helyeként azonosítottak. A beteget 37,5 °C feletti lázzal, mellkasi szorító érzéssel, köhögéssel, fájdalommal és általános gyengeséggel vették föl. Az előzetes etiológiai vizsgálatok többek között az influenzavírusok jelenlétét is kizárták. [Közzétevő: Ha csakugyan etiológiai vizsgálatokkal próbálkoztak egyéb vírusok kizárásával, akkor ez azt jelenti, semmiféle vizsgálatot nem végeztek egyéb vírusok kiszűrésére. Az orvostudományban az etiológia a patológiai betegségek okait, hátterét kutatja. (Wikipédia)] Más gyakori légúti kórokozókat, például humán adenovírusokat is kizártak. Miután a beteg állapota antibiotikum és virális gyógyszerek beadása után sem javult, az intenzív osztályra, majd később egy másik vuhani kórházba szállították további kezelésre. A jelen betegséggel összefüggésbe hozható lehetséges kórokozók azonosítása érdekében a betegtől bronchoalveoláris váladékot (BALF) gyűjtöttek, majd meta-transzkriptomikusan szekvenálták.
A módszerek leírása
A virológiai tudományos publikációk általában tartalmaznak egy módszertani részt, amelyben az eljárást részletesebben ismertetik. Itt ismertetik például a módszertani eljárást és az alkalmazott szekvenálási technológiákat, valamint a bioinformatikai protokollokra vonatkozó információkat.
Adatszolgáltatás
Mint már említettük, pontosan egy személyt vizsgáltak meg. Az elvégzett kísérleteket és az eredmények értékelését nem vakon végezték. Minden érintett tudós teljes körű információval rendelkezett, különösen ismerték a beteg klinikai képét.
Betegtájékoztatás és mintavétel
Egy akut lázzal (37,5 °C feletti hőmérséklet), köhögéssel és mellkasi szorító érzéssel jelentkező beteget vizsgáltak, akit a kínai Vuhani Központi Kórházában vettek fel. A felvétel során a betegtől bronchoalveoláris váladékot (BALF) gyűjtöttek, és a további feldolgozásig -80 °C-on tárolták. Minden beteginformáció, például a vizsgálati és laboratóriumi adatok, klinikai feljegyzések formájában állt rendelkezésre.
A betegminta és az RNS-szekvenálás
Először a teljes RNS-t (ribonukleinsav) kivonták a BALF-ből. Erre a célra kereskedelmi extrakciós készleteket használnak. Ezeket a készleteket általában az adott gyártó utasításai szerint használják. Az extrakciót követően az RNS-oldat mennyiségét és minőségét a későbbi szekvenálás előtt kiértékelték. Mielőtt azonban az RNS-t szekvenálni lehetne, egy úgynevezett RNS-könyvtárat kell létrehozni. Itt a meglévő RNS-t technikailag előkészítik a szekvenálási lépéshez. Az RNS-t például reverz transzkripcióval cDNS-vé kell átírni, és rövid fragmentumokra kell osztani. Jelen esetben a riboszómális RNS-t a könyvtárépítés során a gyártó utasításainak megfelelően távolították el. A riboszomális ribonukleinsav (rRNS) az emberi riboszómákban, a fehérje bioszintézisének helyszínén található. A tényleges szekvenálás előtt a rövid fragmentumokat polimeráz láncreakció (PCR) segítségével felerősítik, hogy a használt szekvenálógép képes legyen detektálni őket és meghatározni a nukleotidszekvenciát. Az RNS-könyvtár szekvenálását az Illumina MiniSeq platformján végezték Sanghajban.
Forrás:[2]
A szekvenciaolvasatok bioinformatikai feldolgozása és a vírus azonosítása.
A fent leírt protokoll szerinti szekvenálás és az azt követő beállítás és minőségellenőrzés a Trimmomatic programmal összesen 56 565 928 rövid, körülbelül 150 nukleotid hosszúságú szekvenciát (olvasatot) eredményezett. A fentieknek megfelelően a rövid szekvenciák a négy nukleotidot jelölő A, T, C, G betűsorozatokkal írhatók le.
A betűsorozatokat közönséges szövegfájlokban tároljuk. Jelen esetben a rövid olvasatokat FASTQ formátumban tároltuk[3], amely további minőségi információkat tartalmaz az egyes nukleotidokra vonatkozóan. Ebből a rövid szekvencián belüli minden egyes nukleotidra vonatkozó minőségi információból meghatározható annak valószínűsége, hogy az adott nukleotidot a szekvenálógép helyesen regisztrálta-e.
| !
info |
Egy FASTQ-fájlban a beolvasott szekvenciák mellett az egyes nukleotidokra vonatkozó minőségi információk is tárolhatók. @3 TACGATGTCEGTTCCGGAAGGTAAGGGEGTGGATGAGACTAAGATCGG +3 FAAAFFFFFFFFFFFFFFFAFFFFF&F6//FFAAFFFF/AFFFFFFF @ : A szekvencia azonosítása + : A szekvencia minőségi információi |
Most kísérletet teszünk arra, hogy a rövid szekvenciaolvasatokból (56 565 928 olvasat x 150 nt hosszú) hosszabb összefüggő nukleotidszekvenciákat határozzunk meg. (nt = nukleotid) Ehhez átfedéseket keresünk a rövid szekvenciák között.
A rövid olvasatok nagy száma miatt – ebben az esetben körülbelül 56 millió – ehhez a lépéshez bioinformatikai programokra és nagy teljesítményű számítógépekre van szükség.
Az átfedéssel számított hosszabb nukleotidszekvenciákat kontignak nevezzük. A kontig szó az angol contiguous szóból származik, ami azt jelenti, hogy összefüggő. A következő ábra szemlélteti ezt a folyamatot.
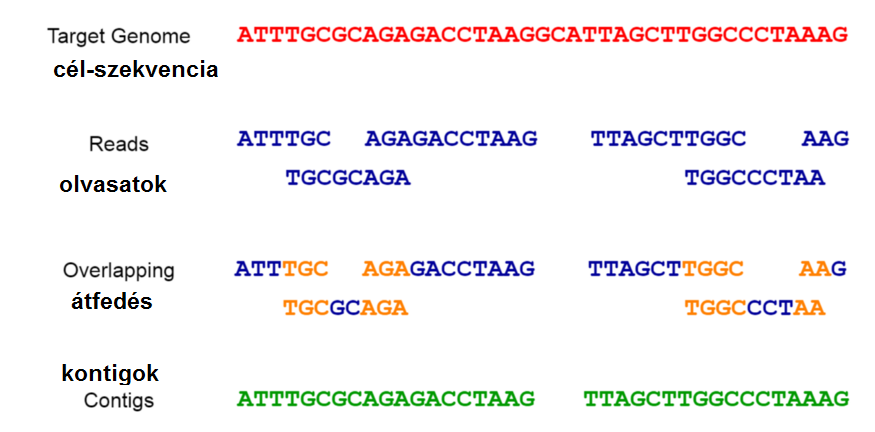
Vegyük észre: A talált kontigok nem fedik le a teljes cél genomot. Ebben az esetben a bioinformatikus a számítógépen tölfi ki a hiányzó rést (gap-filling), azaz a semmiből alkot szoftveres úton a valóságban nem létező nukleinsav-szekvenciákat. A publikációk szerényen hallgatnak róla, mennyi ilyen semmiből teremtett szekvenciát tartalmaz a szoftveres úton alkotot vírus.
Az itt vizsgált publikációban a komplex kontig-generálást a két úgynevezett assemblerrel, a Megahit v.1.1.3 és a Trinity v.2.5.1 verziójával végezték.
Annak érdekében, hogy a szerzők több millió rövid szekvenciájú olvasatból hosszabb kontigokat kapjanak, az alapértelmezett beállításokat használták. Ezt megelőzően azonban a lehetséges emberi eredetű szekvenciákat a humán referencia genom (human release 32, GRCh38.p13)[4] segítségével eltávolították. E lépés után 23 712 657 nem emberi (eddig ismert) rövid szekvencia maradt. A következő táblázat áttekintést ad a két assembler műveletről:
| szoftver | kontigok száma | legrövödebb kontig | leghosszabb kontig |
| Megahit v.1.1.3 | 384 096 | 200 nt | 30 474 nt |
| Trinity v.2.5.1 | 1 329 960 | 201 nt | 11760 nt |
1. táblázat: A Megahit és a Trinity segítségével történő kontiggenerálás eredményeinek áttekintése.
A kontigok összehasonlítása a vonatkozó nukleotid- és fehérjeadatbázisokban található ismert szekvenciákkal nagyfokú hasonlóságot mutatott a SARS-szerű denevérvírus SL-CoVZC45, MG772933-val a leghosszabb kontigok esetében (Megahit: 30 474 nt és Trinity 11 760 nt). A két kontig közül a hosszabbik, 30 474 nukleotid hosszúságú, a szerzők szerint szinte a teljes vírusgenomot lefedte, és azt használták fel az úgynevezett későbbi PCR-megerősítéshez, és a genom jellemzőinek meghatározásához szükséges primerek tervezéséhez.
A vírusgenomot két reprezentatív béta-koronavírus, nevezetesen a SARS-CoV Tor2, AY274119 és a denevér SL-CoVZC45, MG772933 szekvenciaillesztésével rendezték. Az előbbi az emberekkel, az utóbbi a denevérekkel kapcsolatos.
Az adatok elérhetősége
A szekvenciaolvasatokat az NCBI Sequence Read Archive (SRA) adatbázisába töltötték fel a BioProject PRJNA603194 hozzáférési szám alatt. A WHCV (jelenleg SARS-CoV-2) teljes genomszekvenciáját MN908947 azonosító alatt helyezték letétbe a GenBankban.
Kontrollvizsgálatok
„A humán légúti megbetegedésekkel összefüggésbe hozható új coronavírus Kínában” [1] című jelen publikációban nem dokumentáltak kontrollkísérleteket.
________________________________________
2. rész: A módszerek és következtetések kritikai áttekintése
Az állítólagos „új” és „betegséget okozó” SARS-CoV-2 vírus vírusgenomjának meghatározására irányuló eljárás részletes vizsgálatát követően ez a szakasz az egyes lépések kritikai áttekintését tartalmazza. A tudományos ismeretek megkérdőjelezése az élő tudomány elixírje.
1. kritikai megjegyzés: Adatszolgáltatás
Az érintett tudósok nem voltak vakok, ezért ismerték a vizsgált beteg kórtörténetét és tüneteit. Valószínűleg ez befolyásolta a szerzőket abban, hogy a lehetséges légúti kórokozókat keressék. Már ezen a ponton lenne egy egyszerű ellenőrzési lehetőség.
| !
info |
1. Ellenőrzési lehetőség: Egy másik kutatócsoport úgy vizsgálja meg a mintát, hogy nincsenek információi a minta eredetéről, kórképről.
Az ellenőrzés célja: Ugyanazt a vírusgenomot kapja a minta eredetét nem ismerő kutatócsoport is, mint az első? |
________________________________________
2. kritikai megjegyzés: A betegminta és az RNS-szekvenálás
Amint azt a „A betegminta és az RNS-szekvenálás” című részben részletesebben kifejtjük, a tényleges szekvenálás, azaz a rövid fragmentumok nukleotidszekvenciájának meghatározása előtt néhány technikai lépésre van szükség.
Kereskedelmi készleteket használnak például
– a teljes RNS kivonására a mintából,
– a riboszómális RNS eltávolítására a mintából,
– az RNS cDNS-vé alakítására, – az RNS vagy cDNS rövid darabokra történő fragmentálására, vagy
– a cDNS felerősítésére (PCR) a nukleotidszekvencia meghatározása érdekében.
Az RNS mennyiségének és minőségének vizsgálatára kereskedelmi forgalomban kapható technikai berendezéseket használnak. Ezek tehát összetett folyamatok, amelyeket a tudósok csak nagyon korlátozottan tudnak ellenőrizni. Ez közvetlenül elvezet egy másik ellenőrzési lehetőséghez, amelyet a tudományos szakirodalom nem dokumentál.
| !
info |
2. Ellenőrzési lehetőség: A kiindulási próbát egy eltérő protokoll szerint készítik elő a szekvenáláshoz.
Az ellenőrzés célja: Ugyanolyan vagy hasonló olvasatokat kapunk-e eltérő protokollok alkalmazásával? Ezekből ugyanolyan vagy legalább hasonló kontigokat épít-e össze programunk? |
Meg kell tehát jegyezni, hogy a rendelkezésre álló bronchoalveoláris váladékból származó RNS szekvenálása rendkívül összetett protokollt igényel. Az elvégzett lépéseket nehéz ellenőrizni. Ezért kérdéses, hogy a tudományossághoz szükséges reprodukálhatóság mennyire garantált, főleg, hogy nem ismerjük a referencia genomot.
____________________________________
3. kritikai megjegyzés: A szekvenciaolvasatok bioinformatikai feldolgozása és a vírus azonosítása
Először is, szembetűnő a jelentős különbség a két assembler, a Megahit és a Trinitiy eredményei között. Az eltérés óriási. Például a Megahittel való átfedéssel talált leghosszabb összefüggő szekvencia 30 474 nukleotid volt, míg a Trinitiy ugyanebből az adathalmazból 11 760 nukleotid hosszúságú szekvenciát eredményezett. Ezzel szemben a Trinitiy-vel lényegesen több egybefüggő szekvencia-darabot kaptunk, nevezetesen 1 329 960-t, mint a Megahit-tel csak 384.096t.
Itt azonnal felmerül a kérdés:
Milyen lett volna az állítólagos SARS-CoV-2 vírus (program által) javasolt szekvenciája, ha egyrészt a Megahit szoftver, másrészt a megfelelő referencia genomok (denevérszerű SARS-vírusok és korábban ismert, emberrel kapcsolatos SARS-vírusok) nem léteztek volna?
Egy másik fontos kérdés:
Miért csak 29 903 nukleotid[2] hosszú az állítólagos SARS-CoV-2 koronavírus, ami 571 nukleotiddal kevesebb, mint a leghosszabb kontig, amely 30 474 nukleotid?
Rendkívül figyelemre méltó a két szoftver közötti nagy különbség a maximális kontig-hossz tekintetében. Úgy tűnik, nincsenek elismert szabályossági kritériumok, amelyeket az átfedések megtalálására használt algoritmusok esetében követni kellene.
Ez tudományos szempontból különösen kritikus, különösen a reprodukálhatóság szempontjából. Továbbá az eddigi elemzés azt mutatja, különösen a szekvenálási lépés során esetlegesen előforduló véletlen hibák vagy a valóságban esetleg nem létező nukleotidszekvenciák kimutatása tekintetében, hogy elemi logikai megfontolások alapján a kiszámított szekvenciák vagy az állítólagos vírusgenomok csak modellek lehetnek. Ezért nem szabad valódi vírusgenomról vagy valódi vírusszekvenciáról beszélni. Ennek a fontos ténynek a megnevezésével és a modell validálásának lehetséges eljárásaival nem, vagy csak elvétve találkozunk a tudományos irodalomban.
A „Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterising variants” = ” Az assemblerek kiválasztása döntő hatással van a SARS-CoV-2 genom de novo összeállítására és a variánsok jellemzésére”[5] című tanulmányban a szerzők bemutatják, hogy a SARS-CoV-2 genom több száz mintából történő rekonstrukciójában az assembler kiválasztása jelentős szerepet játszik. Kimutatták továbbá, hogy a Megahit és a metaSPAdes (egy másik assembler) által produkált variánsok legalább 9%-ban egyedi módon jellemzőek az alkalmazott assembler módszerre. [Magyarul: Az egyik program ilyen vírusvariánst, a másik olyat alkot ugyanabból a nyersanyagból. ] A tudósok ebből azt a következtetést vonják le [beismerik], hogy a SARS-CoV-2 genom rövid olvasatokból (read) történő összeállításához használt assembler programok döntő szerepet játszanak az úgynevezett variánsok jellemzésében.
Ebből a megállapításból értelemszerűen következik többek között a következő kérdés:
Mik is valójában azok a vírusvariánsok vagy vírusmutációk? Biológiai valóság vagy bioinformatikai alkotás eredménye?
Összefoglalva megállapítható, hogy az assembler eredmények nagy különbségeket mutatnak. A rendelkezésünkre álló publikációban megemlítik, hogy a Megahittel számított leghosszabb összefüggő szekvencia (30 474 nt) szinte a teljes vírusgenomot magában foglalja. E szekvencia alapján tervezték a PCR-primereket. A 8. számú kiegészítő táblázatban [„PCR primers used in this study – „PCR primerek, amelyeket ebben a vizsgálatban használtak”] 52 primert sorolnak fel a genom amplifikációjához. Ezek egyenletesen fedik le a teljes állítólagos vírusgenomot, amint az az alábbi grafikonon látható.
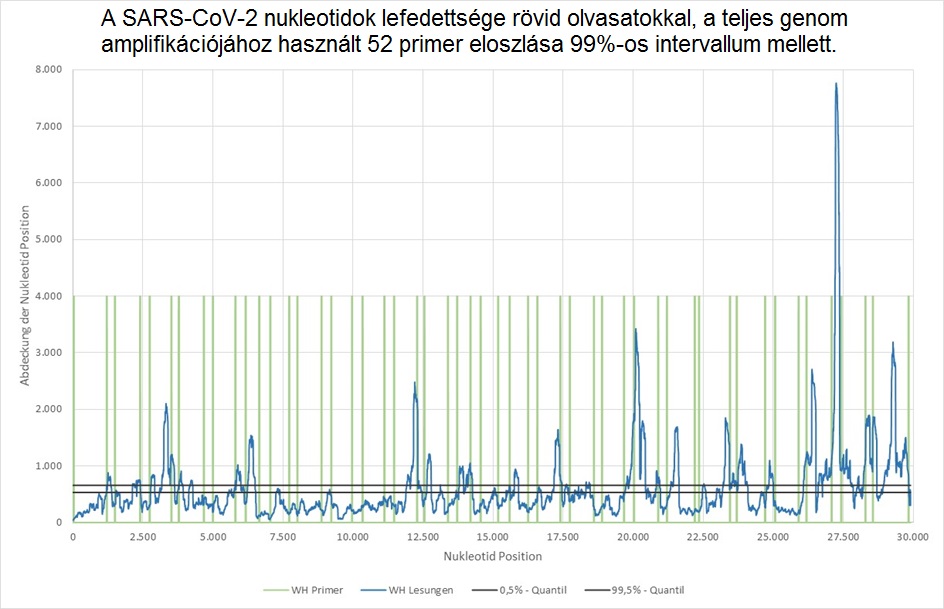
1.ábra: A SARS-CoV-2 nukleotidok lefedettsége rövid olvasatokkal, a teljes genom amplifikációjához használt 52 primer eloszlása 99%-os intervallum mellett. A nukleotidfedettséget a Bowtie2[6] és a Samtools[7] programok segítségével határozták meg.
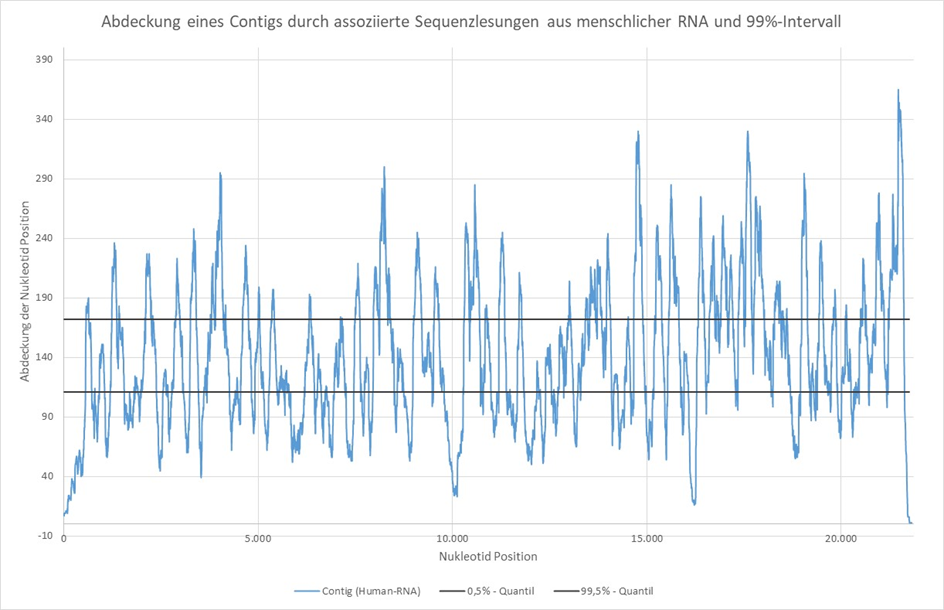
2..ábra: Egy kontig (21 814 nt) lefedettsége a hozzá tartozó szekvenciaolvasatokkal a humán RNS-ből és a 99%-os intervallum mellett. A nukleotidok lefedettségét a Bowtie2[6] és a Samtools[7] segítségével határoztuk meg.
________________________________________
A 2. ábrán látható 21,814 nt hosszúságú kontigot egy kereskedelmi forgalomban kapható humán és nem „fertőzött” sejtkultúra RNS-éből számoltuk ki a Megahit assembler segítségével. A Blastn segítségével végzett összehasonlítás a teljes nukleotid-adatbázissal magas szintű (99,91%-os) egyezést mutatott, szinte a teljes kontig hosszon, a Homo sapiens spectrin repeat containing nuclear envelope protein 2 (SYNE2), transcript variant 5, mRNS, Accession: NM_182914. mRNS-szekvenciával.
A kapcsolódó publikációban „Egy új SYNE2 mutáció, amelyet teljes exom szekvenálással azonosítottak egy koreai Emery-Dreifuss izomdisztrófiás családban” [8]
azt olvassuk az Eredmények alatt
[… novel de novo pathogenic heterozygous missense mutation (NM_182914.2: c.4858G > A; p.Ala1620Thr) of the SYNE2 gene, which had not been previously reported was identified by whole exome sequencing in the proband and by Sanger sequencing in his son. …].
Magyarul:
„… a SYNE2 gén új, korábban nem ismertetett, patogén heterozigóta misszens mutációját (NM_182914.2: c.4858G > A; p.Ala1620Thr) azonosították teljes exom szekvenálással a betegben és Sanger szekvenálással a fiában. …”
Érdekes megfigyelés a maga nemében!
Ettől a megfigyeléstől függetlenül megállapítható, hogy az említett NM_182914.2 azonosítóval rendelkező mRNS-szekvencia összeállítható volt egy nem „fertőzött”, emberi eredetű sejtkultúrából származó RNS-darabkák leolvasásából, amelyeket a tényleges szekvenálás előtt mindössze 14 ciklussal, véletlenszerű és így nem specifikus hexamerekkel amplifikáltak.
A PCR-megerősítés vagy -meghatározás specifikus primerekkel vagy bármilyen referenciaszekvencia használatával nem volt szükséges. Ezért a következő természetes kérdések merülnek fel.
- Miért nem tartalmazza a leghosszabb kontig a SARS-CoV-2 teljes vírusgenom-szekvenciáját?
- Miért van szükség nagyszámú specifikus primer- és referenciaszekvenciára a végső szekvencia-meghatározáshoz?
- Miért hosszabb a leghosszabb kontig (30 474 nt), mint a SARS-CoV-2 állítólagos vírusszekvenciája (29 903 nt)?
Először is, ez ellenkezik az egyszerű logikával. Ha már 30 474 összefüggő nukleotidot találtak, akkor az „igazi” genomszekvenciának biztosan legalább 30 474 nukleotidból kellene állnia.
Ellenkező esetben a Megahit által azonosított átfedések némelyike hibásnak lenne nyilvánítva. A tudományos szakirodalomban erre vonatkozóan nincsenek szabványok dokumentálva. A következőkben egyszerű kontrollkísérletek kerülnek bemutatásra, amelyek csak a Linux operációs rendszer alapszintű ismeretét, a használt bioinformatikai számítógépes programok és a közzétett szekvenciaadatok bizonyos fokú ismeretét igénylik.
| !
info |
1. Kontrollísérlet: A PR]NA603194 BioProject-hozzáférési szám alatt 56.565.928 olvasat található? Protokoll: A fastq-dump –split-files –origfmt –gzip SRR10971381 szekvencia-adatok letöltése |
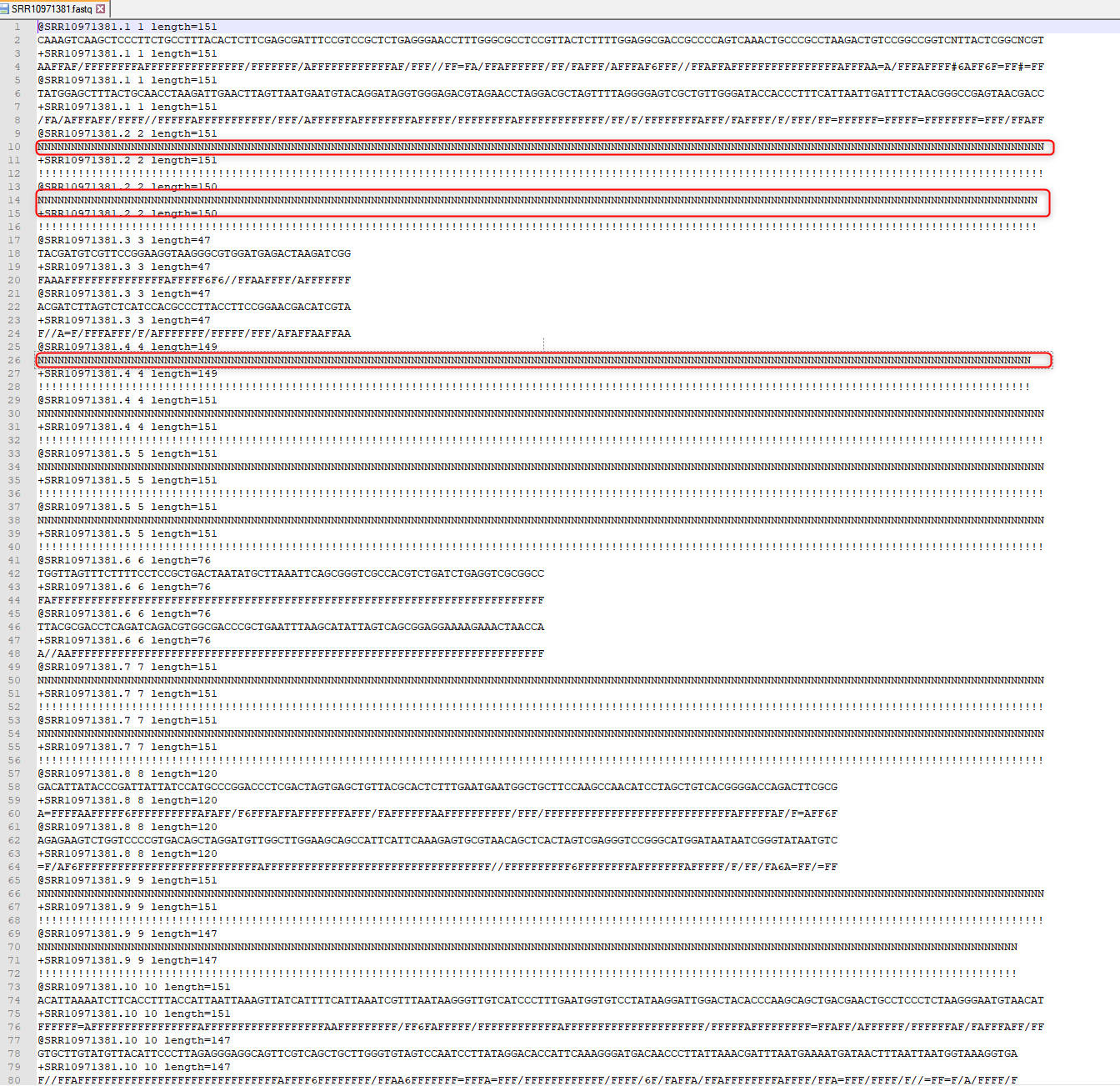
Eredmény: A szekvenciák FASTQ formátumban letölthetők a sratoolkit[9] csomag „fastq-dump” parancsával, és a PairedEnd olvasatok kényelmesen tárolhatók két szöveges fájlban. A két szöveges fájl mindegyike 28 282 964 rövid szekvenciát tartalmaz, amelyek átlagos hossza körülbelül 142 bp.
Összességében tehát mindkét fájl 56 565 928 rövid szekvenciát tartalmaz. Feltűnő azonban, hogy a szekvenciák jelentős része N-ből, azaz ismeretlen nukleotidokból áll.[13] Ezek lehetnek hibás vagy utólag felülírt olvasatok, például emberi szekvenciaolvasatok. A tudományos reprodukálhatóság szempontjából ez az eljárás kritikusnak tekintendő.
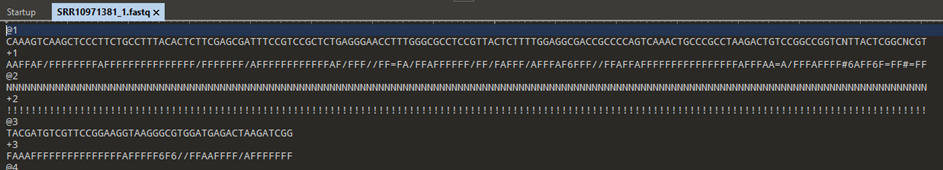
3. ábra: 3 példasorozat az 56 565 928 leolvasásból. A @2 azonosítóval rendelkező olvasat csak ismeretlen „N” nukleotidokból áll, és a legrosszabb minőségi értékelést kapta!
| !
info |
2. kontrollkísérlet: Megismételhetők-e az összeállítások a közzétett szekvenciákkal? Különösen a leghosszabb kontigok reprodukálhatók-e a Megahit és a Trinitiy segítségével? Jegyzőkönyv: Az adatok előkészítése, például az adapterek eltávolítása vagy a rossz minőségű olvasatok eltávolítása az alábbi szoftverekkel: a fastp [10]; fastp -i SRRı10971381_1.fastg.gz -| SRR10971381_2.fastg.gz -o SRR10971381_1.fastq -O SRR10971381_2.fastq Összeállítás (assembly) Megahit v.1.2.9 assemblerrel [11] (alapértelmezett beállítások: megahit -ı SRR10971381_1.fastq -2 SRR10971381_2.fastq -o megahit_result Összeállítás (assembly) a Trinity v.2.5.1 assemblerrel [12] (alapértelmezett beállítások): Trinity –seqType fa –left SRR10971381_1. fa –right SRR10971381_2.fa –CPU 6 –max_memory 20G |
Megjegyzések: Először is meg kell említeni, hogy a Megahit szoftver v.1.1.3 verziója nem futtatható az általunk itt használt számítógépeken. Ezért a jelenlegi v.1.2.9-es verziót használtuk.
A Trinity szoftver nem futtatható FASTQ fájlokkal. Ezért a Trinity-t a SeqType fa (FASTA) beállítással használtuk. A FASTQ fájlokkal ellentétben a FASTA fájlok csak a szekvenciákat tartalmazzák, az egyes bázisokra vonatkozó minőségi információk nélkül. Ezeket a körülményeket figyelembe kell venni az eredmények elemzésekor.
Eredmény: Az alábbi táblázat összefoglalja az eredményeket.
*A fastp szoftver használata nélkül, a közzétett szekvenciákkal végzett összeállítás (assembly) 29 463 kontigot és 29 802 bp hosszúságú leghosszabb kontigot eredményez.
| szoftver | kontigok száma | legrövödebb kontig | leghosszabb kontig |
| Megahit v.1.2.9 | 28 459 | 200 nt | 29 802 nt |
| Trinity v.2.5.1 | 157 283 | 201 nt | 28 875 nt |
2. táblázat: A 2. ellenőrző kísérlet összeállítási eredményei.
A második ellenőrző kísérlet váratlan eredményt mutat. Mind a kontigok teljes száma, mind a maximálisan összefüggő szekvenciák hossza jelentősen eltér a publikált eredményektől.
Szintén figyelemre méltó az a megfigyelés, hogy a Trinitiy-vel hosszabb összefüggő szekvencia számítható ki, mint amilyen a publikációban [1] szerepel. Továbbá a leghosszabb kontigok szinte teljesen megegyeznek az „MN908947” publikált genommal, amint azt a Blastn lekérdezés a standard nukleotid-adatbázisban mutatja.
Így a két leghosszabb kontig egyik esetben sem reprodukálható.
A közzétett szekvenciaadatok tehát nem lehetnek az eredeti, nyers szekvenciaadatok. Figyelemre méltó, hogy a rendelkezésre bocsátott szekvenciák teljes száma (56 565 928) megfelel a vizsgált kiadványban szereplő információknak.
A fent leírtak szerint az emberi eredetű RNS-szekvenciákat kiszűrték. Ehhez a humán referencia genomot (human release 32, GRCh38.p13) használták. A Wikipedia szerint [4]
[… The human reference genome is derived from thirteen anonymous volunteers from Buffalo, New York. Donors were recruited by advertisement in The Buffalo News, on Sunday, March 23, 1997. …].
Magyarul:
„[Az emberi referenciagenom tizenhárom névtelen önkéntesből származik a New York állambeli Buffalóból. Az adományozókat a Buffalo News-ban 1997. március 23-án, vasárnap megjelent hirdetés útján toborozták. …]. ”
Milyen mértékben specifikus a referencia genom az emberre, és ezáltal mennyire alkalmas az emberi eredetű RNS-szekvenciák megbízható felismerésére? Ezt a kérdést a következő kontrollkísérlettel lehet tisztázni, amely természetes módon követi a 2. kontrollkísérletet.
| !
info |
3. ellenőrző kísérlet: A Megahit (v.1.2.9) segítségével a 2. ellenőrző kísérletben kiszámított összefüggő szekvenciák tartalmaznak-e emberi eredetű szekvenciákat? Jegyzőkönyv: A 25 leghosszabb kontig kiválasztása és összehasonlítás az NCBI nukleotid-adatbázisával. Blastn: blastn -db nt -remote -query $Input.fasta -out $result.txt -outfmt „6 qseqid sseqid stitle pident length mismatch gapopen gstart gend sstart send evalue bitscore” |
Eredmény: A következő táblázat a kiválasztott lekérdezési találatokat mutatja. A Megahit segítségével számított leghosszabb kontig (k141_11881) 29 801 nukleotid hosszúságban tökéletes egyezést mutat a SARS-CoV-2 publikált szekvenciájával. A két kontig (k141_18050 és k141_13168) körülbelül 6000 nukleotid hosszúságban nagyfokú egyezést mutat az emberi szekvenciákkal.

- táblázat: A 2. kontrollkísérlet 25 leghosszabb kontigjának és az NCBI nukleotid-adatbázisának összehasonlításából származó kiválasztott lekérdezési találatok.
A tárgyleírások szerint ezek a riboszomális és a hírvivő RNS-ek (rRNS és mRNS). Ez azt jelentené, hogy nem minden emberi RNS-szekvenciát távolítottak el teljes egészében az eredeti szekvenciákból, ahogyan azt a kínai tudósok állították. Végül meg kell jegyezni, hogy az itt vizsgált publikáció 1. és 2. kiegészítő táblázata azt mutatja, hogy a két leghosszabb kontig (Megahit: 30 474 nt és Trinity: 11 760 nt) egyenként 89,1%-os, illetve 90,4%-os nukleotidonkénti egyezést mutat a denevérkoronavírus SL-CoVZC45, MG772933 denevérrel. Azt azonban nem közlik, hogy összesen hány nukleotid egyezik. Nagyon is lehetséges, hogy a Megahit esetében az összes 30 474 nukleotidból csak 10 000 nukleotidra van egyezés, 89,1%-os arányban. Ez azt jelentené, hogy 20 474 nukleotidnak nincs jelentős egyezése a denevér SL-CoVZC45-vel.
________________________________________
4. kritikai megjegyzés: Kontrollkísérletek
Mint már említettük, az itt vizsgált kiadványban nem dokumentáltak kontrollkísérleteket. Ezért feltételezhető, hogy nem végeztek kontrollkísérleteket.
A következőkben további nyilvánvaló ellenőrzési lehetőségeket ismertetünk, amelyek biztosíthatták volna a kapott eredményeket.
| !
info |
3. Ellenőrzési lehetőség: A megállapított vírusgenom nyerhető-e más, azonos tünetegyüttesű megbetegedésekből származó mintákból is, viszon egészsége semberekből származó mintákból nem.
Ellenőrzési cél: Annak a feltételezésnek a megerősítése, hogy a kiszámított szekvencia jelenléte ok-okozati összefüggésben áll egy adott tünetegyüttessel, lehet-e (társ)felelős egy nagyon specifikus tünetegyüttesért. 4. ellenőrzési lehetőség: |
________________________________________
Összefoglalás és következtetés
Egy Vuhanban történt megbetegedésből származó betegmintából a teljes RNS-t extrahálták. A szerzők szerint a folyamat során emberi RNS-fragmenteket eltávolították.
- A 3. kontrollkísérlet megmutatta, hogy az MN908947 azonosítóval ellátott állítólagos SARS-CoV-2 vírusgenom közzétett szekvenciáiban nagy valószínűséggel még mindig jelen van emberi eredetű riboszómális vagy hírvivő RNS, feltéve, hogy az adatbázisokat megfelelően karbantartják.
- A 2. ellenőrző kísérlet azt mutatta, hogy a közzétett eredmények nem reprodukálhatók. Sokkal valószínűbb, hogy a közzétett szekvenciák nem azonosak az eredeti szekvenciákkal.
- A jelen publikációban szereplő leírás alapján nem feltételezhető, hogy a Megahit és a Trinitiy segítségével megkapható a csaknem a teljes „vírusgenom”. Ez a megállapítás azért aggályos, mert már nem lehet ellenőrizni, hogy a Megahit segítségével összeállított 30 474 nt hosszúságú kontig milyen mértékben egyezik meg a bat SL-CoVZC45 denevér koronavírussal, és hogy ez milyen mértékben tér el a SARS-CoV-2 végleges szekvenciajavaslatától.
- Nem dokumentáltak kontrollkísérleteket. Így feltételezhető, hogy nem végeztek kontrollkísérleteket. Felvázoltuk a lehetséges ellenőrző kísérleteket, de a lista nem teljes.
- A priori teljesen tisztázatlan, hogy a vizsgált beteg betegsége „vírusos” eredetű-e.
- Ezen túlmenően bebizonyosodott, hogy az állítólagos vírusgenom létrehozásához használt RNS-darabkák eredetét nem állapították meg. Lehet, hogy egyszerűen emberi eredetű RNS.
- Felmerül az a kérdés is, hogy az itt alkalmazott módszerekkel egyáltalán lehetséges-e tisztázni az eredetet. Röviden: a felhasznált szekvenciák „vírusos” eredetűek vagy sem? Ez a kérdés elkerülhetetlenül elvezet a vírusizoláció fogalmához, az „izoláció” szó értelmében. Jelen esetben nem találtak (vírusos) részecskét.
Egy lehetséges (vírusos) részecske megtalálására a következő egyszerű protokoll lehetne célravezető:
1, Keressünk „sok” azonos morfológiájú részecskét a beteg mintájában (itt: bronchoalveoláris váladék). Ezeknek létezniük kell, mivel a virológusok az állítólagos SARS-CoV-2 vírusrészecskék méretét körülbelül 100 nm-es tartományba sorolják.
2, Válasszuk el ezeket a részecskéket minden mástól.
3, Vegyünk több különböző mintát a talált részecskékből.
4, Minden mintát egymástól függetlenül szekvenáljunk.
5, Amennyiben minden esetben (majdnem) ugyanazt a szekvenciát kapjuk:
Akkor tudományos értelemben egy olyan részecskét írhatunk le, amelyet szekvenciája és morfológiája jellemez. A részecske esetleges patogenitásának vagy átvihetőségének (fertőzőképességének) bizonyítéka azonban még messze nem állna rendelkezésünkre.
Összefoglalva tehát ki kell jelenteni:
Az „A new coronavirus associated with human respiratory disease in China”[1] című publikáció nem bizonyította „új vírus”, új „vírusszekvencia” vagy kórokozó létezését. Különösen a vizsgált beteg klinikai tünetegyüttesére nem találtak ok-okozati összefüggést.
Hivatkozások
[1] A new coronavirus associated with human respiratory disease in China | Nature volume 579, pages 265–269 (2020)
[2] Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co – Nucleotide – NCBI (nih.gov)
Súlyos akut légzőszervi szindrómás koronavírus 2 izolátum Vuhan-Hu-1, co – Nukleotid – NCBI (nih.gov)
[3] FASTQ formátum. 2021 augusztus: https://en.wikipedia.org/wiki/FASTQ_format
[4] Referencia genom. 2021. szept.
link: https://en.wikipedia.org/wiki/Referencia_genom
[5] Rashedul Islam et al. ” Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants – PubMed (nih.gov). In: Briefings in Bioinformatics 22.5 (2021). DOI: 10.1093/bib/bbab102.
Az assemblerek kiválasztása döntő hatással van a SARS-CoV-2 genom de novo összeállítására és a variánsok jellemzésére – PubMed (nih.gov).
[6] Bowtie 2: fast and sensitive read alignment (sourceforge.net): (Bowtie 2: gyors és érzékeny olvasás összehangolás)
[7] GitHub – samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data Eszközök (C nyelven, htslib használatával írva) az újgenerációs szekvenálási adatok manipulálására.
[8] A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy – ScienceDirect In: Clinica Chimica Acta 506 (2020), pp. 50-54. DOI: 10.1016/j.cca.2020.03.021.
(Teljes exom szekvenálással azonosított új SYNE2 mutáció egy koreai Emery-Dreifuss izomdisztrófiás családban)
[9] GitHub – ncbi/sra-tools: SRA Tools
[10] GitHub – OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/filtering/splitting/merging…)
(Egy ultragyors, minden egyben FASTQ előfeldolgozó (QC/adapterek/vágás/_szűrés/osztás/összevonás…)
[11] https://github.com/voutcn/megahit.
Voutcn. voutcn/megahit: Ultragyors és memória-hatékony (meta-)genom-assembler.
[12] https://github.com/trinityrnaseq/trinityrnaseq
[13] Utasítások a szekvenciák letöltéséhez, hogy láthassa a feketére festett („N” betűvel helyettesített) szekvenciákat.
1. töltse le a szekvenciákat fastq-dump segítségével (konzolprogram Windows vagy Linux operációs rendszerre).
fastq-dump –split-files –origfmt –gzip SRR10971381
2. ha csak egy részt szeretne letölteni (1-től 100-ig, azaz 100 olvasatot:
fastq-dump –split-files -N 1 -X 100 -Z SRR10971381 > SRR10971381.fastq
3. Itt a hivatalos link https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR10971381
Az első 100 párosított végű leolvasás, amelyet a 2. pont alatti paranccsal hoztunk létre. Még az első 100 között is számos olvasat feketére van festve.
[14] Kontrollexperiment Phase 1 – Mehrere Labore bestätigen die Widerlegung der Virologie durch den cytopathischen Effekt | [Telegraph]
(Ellenőrző kísérlet 1. fázis: – több laboratórium megerősíti a hivatalos virológia cáfolatát a citopátiás hatás kérdésében).
Táblázat
[8. táblázat]: 41586_2020_2008_MOESM1_ESM.pdf (springer.com)
2022. augusztus
Samuel Eckert írása – Kontrollexperiment Phase 2 – Entlarvt: Wie in der maßgeblichen Studie zu SARS-CoV-2 durch die chinesischen Wissenschaftler getrickst wurde – Telegraph – alapján közzéteszi
Király József

Köszönöm, kedves József, hogy nem hagyja kihunyni a lángot és az újabb nagyszerű publikáció! Baráti üdvözlettel